Fibrosi cistica
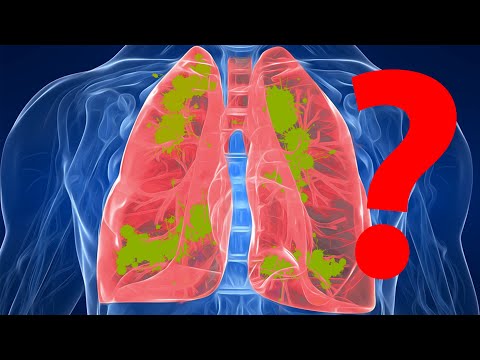
A fibrosi cistica hè una malattia chì face cresce mucosi spessi è appiccicosi in i pulmoni, in u trattu digestivu è in altre zone di u corpu. Hè una di e malatie pulmonari croniche più cumune in i zitelli è i ghjovani adulti. Hè un disordine chì mette in periculu a vita.
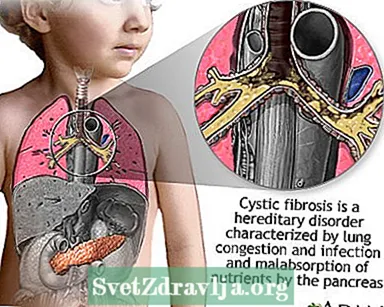
A fibrosi cistica (CF) hè una malattia chì si trasmette cù e famiglie. Hè causatu da un genu difettu chì face u corpu pruduce un fluidu anormalmente densu è appiccicosu, chjamatu mucus. Stu mucus s'accumula in i passaghji respiratorii di i pulmoni è in u pancreas.
L'accumulazione di mucus risulta in infezioni pulmonari periculose per a vita è serii prublemi di digestione. A malatia pò ancu influenzà e ghiandole sudoripare è u sistema riproduttivu di l'omu.
Parechje persone portanu un genu CF, ma ùn anu micca sintomi. Hè perchè una persona cun CF deve eredità 2 geni difettosi, 1 da ogni genitore. Alcuni americani anu u genu CF. Hè più cumunu trà quelli di discendenza nordu o centrale europea.
A maiò parte di i zitelli cun CF sò diagnosticati da l'età di 2 anni, soprattuttu chì u screening di u novu hè fattu in i Stati Uniti. Per un picculu numeru, a malatia ùn hè micca rilevata finu à l'età di 18 anni o più. Questi zitelli anu spessu una forma più dolce di a malattia.
I sintomi in i neonati ponu include:
- A crescita ritardata
- Fallimentu di guadagnà pesu normalmente durante a zitiddina
- Nisun muvimentu intestinale in e prime 24 à 48 ore di vita
- Pelli à gustu salitu
I sintomi relativi à a funzione intestinale ponu include:
- Dolore di panza da stinzia severa
- Aumentazione di gas, gonfiore, o una pancia chì pare gonfia (distesa)
- Nausea è perdita di l'appetite
- Sgabelli chì sò pallidi o argillosi, puzzulanti, anu mucus, o chì flottanu
- Perda di pesu
I sintomi relativi à i pulmoni è i sinus ponu include:
- Tosse o aumentu di mucus in i sinus o pulmoni
- Stanchezza
- Congestione nasale causata da i polipi nasali
- Episodi ripetuti di pulmonite (sintomi di pneumonia in qualcunu cù fibrosi cistica includenu febbre, tosse aumentata è mancanza di respiru, mucus aumentatu è perdita di appetitu)
- Dolore sinusale o pressione causata da infezzione o polipi
Sintomi chì ponu esse rimarcati più tardi in a vita:
- Infertilità (in l'omi)
- Infiammazione ripetuta di u pancreas (pancreatite)
- Sintomi respiratorii
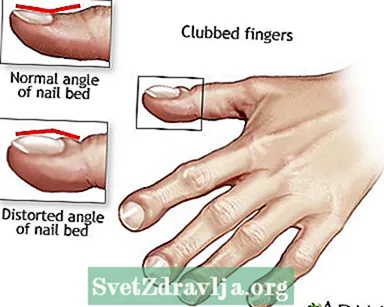
- Dita sbattute
Un test di sangue hè fattu per aiutà à rilevà CF. U test cerca cambiamenti in u genu CF. Altri testi aduprati per diagnosticà CF includenu:
- U test di tripsinogenu immunoreattivu (IRT) hè un test di screening standard di u neonatu per CF. Un altu livellu di IRT suggerisce possibili CF è richiede ulteriori test.
- U test di cloruru di sudore hè u test di diagnosticu standard per CF. Un altu nivellu di sale in u sudore di a persona hè un segnu di a malatia.
Altri testi chì identificanu i prublemi chì ponu esse ligati à CF includenu:
- Radiografia di u torace o CT scan
- Test di grassu fecale
- Prove di funzione pulmonare
- Misurazione di a funzione pancreatica (feci elastase pancreatica)
- Test di stimulazione secretina
- Tripsina è chimotripsina in feci
- GI superiore è serie intestinali teneri
- Culture pulmonari (ottenute da sputum, bronchoscopia o tampone di gola)
Un diagnosticu precoce di CF è un pianu di trattamentu pò migliurà a sopravvivenza è a qualità di vita. U seguitu è u seguitu sò assai impurtanti. Quandu hè pussibule, a cura deve esse ricevuta in una clinica di specialità di fibrosi cistica. Quandu i zitelli ghjunghjenu à l'età adulta, devenu trasferì in un centru di specialità di fibrosi cistica per adulti.
U trattamentu per i prublemi pulmonari include:
- Antibiotici per prevene è trattà infezioni pulmonari è sinusali. Pò esse presi per bocca, o dati in e vene o cù trattamenti respiratorii. E persone cun CF ponu piglià antibiotici solu quandu ci vole, o tuttu u tempu. E dosi sò spessu più alte di u normale.
- Medici inalati per aiutà à apre e vie aeree.
- Altri medicini chì sò dati da un trattamentu respiratoriu per diluisce u mucus è facenu più faciule per tossisce sò a terapia enzimatica DNAse è e soluzioni saline altamente cuncintrate (salina ipertonica).
- Vaccinu contra a gripe è vaccinu contra u polisaccharide pneumococcal (PPV) annu (dumandate à u vostru duttore di salute).
- U trapianto di pulmone hè una opzione in certi casi.
- A terapia di l'ossigenu pò esse necessaria postu chì a malatia pulmonaria s'aggrava.
I prublemi pulmonari sò ancu trattati cù terapie per diluisce u mucus. Questu facilita a toscia di u mucus fora di i pulmoni.
Questi metudi includenu:
- Attività o eserciziu chì vi face respira prufondu
- Dispositivi chì sò usati durante u ghjornu per aiutà à liberà e vie aeree di troppu mucus
- Percussione toracica manuale (o fisioterapia toracica), in cui un membru di a famiglia o un terapeuta batte leggermente u pettu, a schiena è a zona di a persona sottu à e bracce
U trattamentu per l'intestini è i prublemi nutrizionali pò include:
- Una dieta speciale ricca di proteine è calorie per i zitelli maiò è l'adulti
- Enzimi pancreatici per aiutà à assorbe i grassi è e proteine, chì sò presi cù ogni pastu
- Integratori di vitamine, in particulare vitamine A, D, E è K
- U vostru fornitore pò cunsiglià altri trattamenti se avete feci assai duri
Ivacaftor, lumacaftor, tezacaftor è elexacaftor sò medicinali chì trattanu certi tipi di CF.
- Miglioranu a funzione di unu di i geni difettosi chì causanu CF.
- Finu à 90% di i pazienti cun CF è ammissibili à unu o più di sti medicinali solu o in cumbinazione.
- Di conseguenza, ci hè menu accumulazione di mucus spessi in i pulmoni. Altri sintomi CF sò migliurati dinò.
A cura è a sorveglianza in casa duveranu include:
- Evitendu u fume, a polvera, a terra, i fumi, i chimichi di a casa, u fumu di u caminu, è a muffa o a muffa.
- Dà assai liquidi, in particulare à i zitelli è zitelli in tempu caldu, quandu ci hè diarrea o sgabelli sciolti, o durante attività fisica in più.
- Eserciziu 2 o 3 volte à settimana. U nuatu, u jogging è u ciclismu sò bone opzioni.
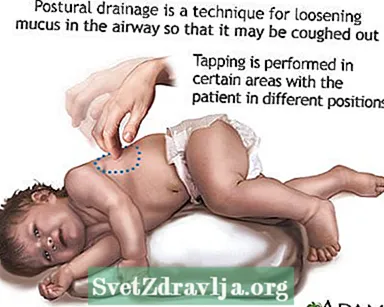
- Scaricamentu o purtamentu di mucus o secrezioni da e vie aeree. Questu deve esse fattu da 1 à 4 volte ogni ghjornu. I pazienti, famiglie è caregivers devenu amparà à fà percussione toracica è drenaje posturale per aiutà à tene libere e vie aeree.
- Nisun cuntattu cù altre persone cun CF hè cunsigliatu perchè ponu scambià infezioni (ùn vale micca per i membri di a famiglia).
Pudete calmà u stress di a malattia unendu si à un gruppu di supportu per a fibrosi cistica. A spartera cù l'altri chì anu sperienze è prublemi cumuni pò aiutà a vostra famiglia à ùn sentesi micca sola.
A maiò parte di i zitelli cun CF stanu in bona salute finu à ghjunghje à l'età adulta. Sò capace di participà à a maiò parte di l'attività è di frequentà a scola. Parechji ghjovani adulti cù CF finiscenu l'università o trovanu impiegu.
A malatia pulmonaria si aggrava finu à u puntu induve a persona hè disattivata. Oghje, a durata media di vita per e persone cun CF chì campanu finu à l'età adulta hè di circa 44 anni.
A morte hè più spessu causata da complicazioni pulmonari.
A cumplicazione più cumuna hè l'infezzione respiratoria cronica.
Altre complicazioni includenu:
- Problemi intestinali, cum'è i calcoli biliari, u bloccu intestinale è u prolapsu rettale
- Tussendu sangue
- Fallimentu respiratoriu crònicu
- Diabetes
- Infertilità
- Malatie di u fegatu o fallimentu di u fegatu, pancreatite, cirrhosi biliare
- Malnutrizione
- Polipi nasali è sinusite
- Osteoporosi è artrite
- Pneumonia chì volta à turnà
- Pneumotorace
- Fallimentu cardiacu di u latu dirittu (cor pulmonale)
- Cancer culettale
Chjamate u vostru duttore se un criaturu o un zitellu hà sintomi di CF, è sperienze:
- Febbre, tosse aumentata, cambiamenti in sputum o sangue in sputum, perdita di appetitu, o altri segni di pneumonia
- Aumentata a perdita di pesu
- Sintomi o feci più frequenti chì puzzulanu o anu più mucus
- Ventre gonfiu o gonfiore aumentatu
Chjamate u vostru duttore se una persona cun CF sviluppa novi sintomi o se i sintomi aggravanu, in particulare difficultà respiratoria severa o tossisce sangue.
CF ùn pò esse impeditu. U screening di quelli chì anu una storia di famiglia di a malattia pò rilevà u genu CF in parechji trasportatori.
CF
- Nutrizione enterale - zitellu - gestione di i prublemi
- Tubu d'alimentazione di gastrostomia - bolus
- Cume respira quandu vi manca u fiatu
- Tubu d'alimentazione di Ghjejunostomia
- Drenaggio posturale
 Clubbing
Clubbing Drenaggio posturale
Drenaggio posturale Dita sbattute
Dita sbattute Fibrosi cistica
Fibrosi cistica
Donaldson SH, Pilewski JM, Griese M, et al. Tezacaftor / ivacaftor in sughjetti cù fibrosi cistica è F508del / F508del-CFTR o F508del / G551D-CFTR. Am J Respir Crit Care Med. 2018; 197 (2): 214-224. PMID: 28930490 pubmed.ncbi.nlm.nih.gov/28930490/.
Eagan ME, Schechter MS, Voynow JA. Fibrosi cistica. In: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Manuale di Nelson di Pediatria. 21a ed. Filadelfia, PA: Elsevier; 2020: chap 432.
Farrell PM, White TB, Ren CL, et al. Diagnosticu di fibrosi cistica: linee guida di consensu da a Fundazione di Fibrosi Cistica. J Pediatr. 2017; 181S: S4-S15.e1. PMID: 28129811 pubmed.ncbi.nlm.nih.gov/28129811/.
Graeber SY, Dopfer C, Naehrlich L, et al. Effetti di a terapia lumacaftor / ivacaftor nantu à a funzione CFTR in i pazienti omosigoti Phe508del cù fibrosi cistica. Am J Respir Crit Care Med. 2018; 197 (11): 1433-1442. PMID: 29327948 pubmed.ncbi.nlm.nih.gov/29327948/.
Grasemann H. Fibrosi cistica. In: Goldman L, Schafer AI, eds. Medicina Goldman-Cecil. 26a ed. Filadelfia, PA: Elsevier; 2020: chap 83.
Rowe SM, Hoover W, Solomon GM, Sorscher EJ. Fibrosi cistica. In: Broaddus VC, Mason RJ, Ernst JD, et al, eds. Manuale di Murray è Nadel di Medicina Respiratoria. 6a ed. Filadelfia, PA: Elsevier Saunders; 2016: chap 47.
Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor-ivacaftor in pazienti cun fibrosi cistica omozigote per phe508del. N Engl J Med. 2017; 377 (21): 2013-2023. PMID: 29099344 pubmed.ncbi.nlm.nih.gov/29099344/.
Cunsigliatu

Identificà è Trattà Eruzione di Denti
